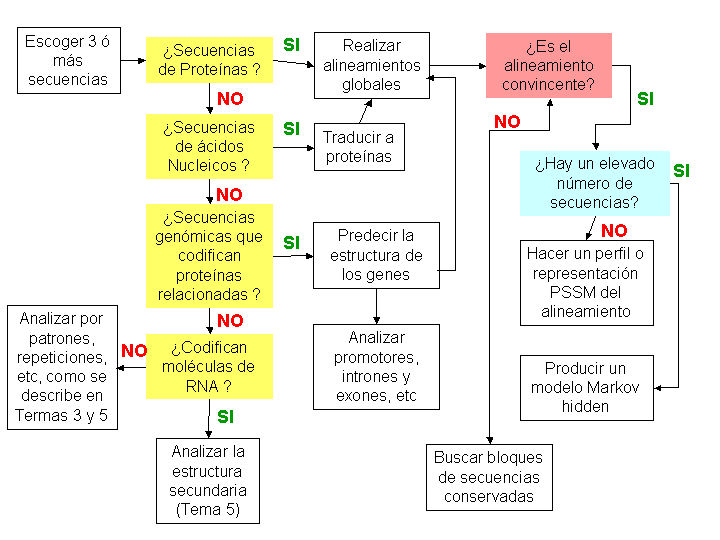
Métodos para el alineamiento de secuencias de proteínas y ácidos nucleicos
- Mediante el alineamiento
global progresivo de las secuencias, también llamado
de programación dinámica. Con este método, las
primeras secuencias que se disponen en el alineamiento son las que más se parecen entre si y se colocan por ello en primer lugar. Luego se va construyendo un alineamiento
con el resto de las secuencias que se van incorporando ajustándose a las dos primeras. Algunos programas
que usan este método son:
- La serie de programas CLUSTAL, con mucho los más conocidos
- MUSCLE (porque es muy rápido)
- TCOFFE (porque es muy preciso. Permite combinar secuencias y estructuras, buscar en la base de datos PDB, etc..)
- programas como TREECON para hacer bootstrapping con secuencias alineadas
- Estos programas, y
en particular Clustal realizan comparaciones de todas las
secuencias entre sí usando el algoritmo de alineamiento global de Needleman-Wunsch, siendo capaces de crear secuencias consensos.
- El problema que puede
surgir con este tipo de alineamientos, es que dependen del alineamiento
de las dos secuencias mas semejantes entre si. Si se comete un error con
ellas, el error se traslada al resto de las secuencias que se incoporan
al proyecto de secuenciación. Estos errores son más
problemáticos cuando las primeras secuencias, las más
semejantes entre si, y son dispares entre sí.
- Estos métodos
de alineamientos son magnificos cuando las secuencias son similares
entre sí
- El programa Clustal
está especialmenbte diseñado para dar un buen alineamientos
con un número elevado de secuencias, dando una indicación
muy buena de la estructura en dominios de estas secuencias que se
comparan.
- Criterios para la elección de secuencias
- Criterios generales
- ¿Extracción de secuencias?
- Análisis de las anotaciones para buscar fuentes de variaciones
- Alternativas cuando tus comparaciones no son lo suficientemente buenas (buscadores de dominios):
- El servicio Pratt (para identificar motivos en tus secuencias usando PROSITE)
- Otros servidores que realizan busquedas como:
- eMotifSearch y eMotifScan (que deben instalarse localmente en algunos casos)
- MDScan para buscar lugares de unión de factores de transcripción
- 3motif para visualizar la estructura tridimensional de motivos encontrados dentro de PDB
- eMOTIF Search para buscar motivos conservados dentro de las proteínas
- El servidor MEME para localizar dominios conservados dentro de un conjunto de proteínas
- El servidor Improbizer para encontrar sitios de unión de proteínas al ADN y al ARN
- El servidor Motif Matcher para buscar motivos dentro de tus proteínas
|
{kind=link}