TUTORIAL GENÓMICA BACTERIANA
ENSAMBLADO DE GENOMA DE E. coli
- Descarga de los archivos de secuencias
- Análisis de la calidad de las secuencias con FastQC
- Pre-análisis con el uso de comandos bash para comprobar el número de secuencias, si las secuencias son pareadas y si están sincronizadas
- Pre-procesado de secuencias con programas adecuados para el filtrado de secuencias (de ser necesario)
- Descarga y lectura del manual de Velvet. Instalación de Velvet (método avanzado)
- Ensamblado de secuencias pareadas con Velvet.
- Análisis del valor de k-mer mas adecuado para realizar el ensamblado: introducción al valor de N50.
- Alineamiento y comparación de los ensamblados obtenidos con diferentes valor de K comparándo con un genoma de referencia de calidad (trusted genome) con ayuda del programa Mauve
PASOS A DAR..
1. Obtención de las secuencias
Estas prácticas dependen de que hayamos secuenciados el genoma de una bacteria, en este caso E.coli. O bien lo hacemos realmente usando un servicio de secuenciación comercial, o bien obtenemos los datos desde las páginas públicas en Internet, como las de ENA en el EMBL
- Hasta hace unos dias, las secuencias obtenidas mediante un equipo Illumina estaban depositadas en el ENA (European Nucleotide Archive) del EBI (European Bioinformatic Institute). Desde allí se podían descargar las secuencias de E.coli O14:H4 buscando en la página WEB por los datos SRR292770.
- Sin embargo, ahora el autor ha retirado esas secuencias de la base de datos públicas, pero una búsqueda en Google con SRR29770 localiza estos mismos datos de secuenciación en la base de datos de Japón DDBJ.
- Puedes buscar esos datos con Google y luego hacer la descarga. Y verás que hay dos archivos, llamados SRR29770_1.fastq.bz2 y SRR29770_2.fastq.bz2, que están comprimidos en el formato bz2, un formato de compresión que no está en un principio soportado por Velvet, el ensamblador que vamos a usar.
- (opcional) Lo que vamos a hacer es convertirlos al formato gz, que si es un formato compatible con Velvet. Como parte de la práctica, deberías encontrar información con Google sobre un programa que te permita descomprimir los archivos en formato bz2, y luego otro programa que creara los archivos comprimidos de nuevo en formato gz. Así dejamos satisfechos a Velvet, que puede trabajar con archivos comprimidos para no tener que usar mucho espacio en nuestras cuentas
- Cuando este archivo era accesible en el ENA, pude ver que se corresponden a secuencias pareadas hechas con un equipo Illumina HiSeq2000 con ADN obtenido de la estirpe de E.Coli TY-2482.
- Si no quieres jugar y aprender a descomprimir y volver a comprimir los archivos, puedes descargartelo ya directamente desde los enlaces siguientes que se encuentran en mi cuenta personas de Mega. Puedes optar por descargar los archivos en tu ordenador, o hacer una copia de ellos en tu cuenta de Mega (si te registras, te dan grátis 50Gb para almacenar datos)
2. Preprocesado y análisis preliminar de las secuencias (Esto es una repetición de la instalación del programa FastQC y del análisis de la secuencias que ya ha sido tratada en la práctica anterior). Puedes optar por leer para recordar lo que hay que hacer, o simplemente analizar los datos directamente con FastQC.
- Es absolutamente necesario valorar si es necesario pre-procesar las secuencias (filtrarlas por criterios de calidad) antes de
realizar un ensamblado. El pre-procesado implica:
- Mirar con comandos linux si estas secuencias son simples o pareadas. Si contienen ambos archivos el mismo número de secuencias, y si estos dos archivos están sincronizados o no (esto significa que no solo hay el mismo número de secuencias, sino que el orden en el que están las lecturas es idéntico.
- De ser necesario, elimina mediante un filtrado las secuencias de mala calidad, como aquellas mal secuenciadas (secuencias llenas de “N”), aquellas que son más cortas de lo debido.
- Eliminar la redundancia en la secuenciación y la duplicación de secuencias (especialmente en el caso del ensamblado de secuencias genómicas)
- Valorar y eliminar las secuencias de los adaptadores si es que están presentes y descritas en el apartao de secuencias sobrerepresentadas
- El programa fastqc, aunque se puede usar en un entorno gráfico que se agradece, debería ser usado en el modo interactivo de comandos (nada de ventanitas) porque luego habrá que usarlo en superordenadores que no aceptarán el modo gráfico de Java. Para hacerlo solo tienes que ejecutar fastqc e incluir el nombre del archivo que quieres procesar. Por ejemplo: fastqc Sec_Illumina.fastq.gz. Verás que se crea una serie de archivos entre los que se incluye unos html que corresponden a páginas WEB. Podrás descargártelas con programas como Filezilla (versión cliente) o a través de una versión de FTP (File Transfer Protocol) implementado desde Linux. Puedes obtener información desde estas páginas https://ubunlog.com/comando-ftp-uso-basico/. Si conoces la dirección absoluta de ese archivo puedes hasta usar el comando wget.
Probemos a descargar e instalar FastQC. Entro en la página http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- También es posible descargar e instalar el programa desde el
Centro de Software de Ubuntu. Para ello, pulsamos sobre el icono
 de
Ubuntu, y en el campo donde está la lupa, escribimos fastqc. La ventaja que
tenemos en este caso, es que fastqc se instala automáticamente y se puede
ejecutar sin más desde cualquier ventana de comandos de nuestro Linux, porque la localización del programa se indica en el PATH.
de
Ubuntu, y en el campo donde está la lupa, escribimos fastqc. La ventaja que
tenemos en este caso, es que fastqc se instala automáticamente y se puede
ejecutar sin más desde cualquier ventana de comandos de nuestro Linux, porque la localización del programa se indica en el PATH. - Y
descargo el programa. Si no funciona (ya sea en Windows o en Linux), es que probablemente
no se tiene bien instalado el Java. Y es que este programa necesita la
instalación del Java Development Kit, no el java normal.
- Para ver si tienes instalado Java, teclea java -version
- Para instalar la versión JDK de Java, busca con Google cómo se hace en Linux
- Con el Java JDK instalado, programas como FastQC, Mauve, etc, se
abrirán como programas que se abren en un entorno gráfico (con ventanas y con
menús a los que se puede acceder con ayuda del ratón).
- NOTA 1: No todos los programas jar de JAVA se ejecutan como una ventana en entorno gráfico. Por ejemplo, se podrá usar las herramientas Picard (para manipular secuencias SAM) pero solo en modo de comandos desde una ventana de Terminal (no se abrirá nunca una ventana gráfica que nos permita usar el programa)
- NOTA 2: Algunos programas, como FastQC, también se pueden ejecutar desde una ventana terminal con comandos, algo que es interesante si estás trabajando en un superordenador en el que no puedes tener acceso a un modo gráfico
- Puedes investigar (si quieres aprender) la exsitencia de una plataforma WEB llamada Galaxy en el que está instalado FastQC y muchos otros programas. Pero hay que tener en cuenta de estaremos utilizando servidores “prestados” que tiene las limitaciones que impone el servidor donde quiera que esté alojado. Por ejemplo, tendremos un máximo de 2Gb para la descarga y generación de datos en muchos casos, y lo mismo con una secuenciación masiva tienes muchos Tb de datos. Además, Galaxy no se puede usar en los supercomputadores que hay implementado para nuestro propio uso, ya que éstos carecen de entornos gráficos. Por eso, no lo enseñamos en clase.
- Si el programa no te funcionaba, es que no tenías el Java instalado o bien instalado.
- En Windows lo lanzo FastQC o bien haciendo un doble clic sobre el archivo run_fastqc.bat o bien seleccionas este archivo y le das con el botón derecho del ratón. Se selecciona la opción Abrir y se indica la opción Java JDK.
- Con Linux, nos desplazamos al directorio donde hemos instalado FastQC (con la orden $cd nombredirectorio) y siguiendo las instrucciones de instalación que hay que leerse y que está en la carpeta o directorio del propio programa, hay que activar un archivo lanzador llamado fastqc cambiando los permisos con la orden chmod (change mode). Esta orden cambia los permisos de lectura y/o escritura y/o de ejecución de los archivos en Linux. Es necesario tener permisos de root o de usuario en su cuenta para poder hacerlo. Para que un programa pueda funcionar como tal, es necesario hacerlo ejecutable y muchas veces los programas que nos descargamos para Linux no son ejecutables hasta que el usuario lo convierta en tal. Para cambiar los permisos escribimos:
- $sudo chmod 755 fastqc (dar el password si lo pregunta)
- Alternativamente se puede escribir chmod +x fastqc
- Se puede comprobar cómo hemos cambiado los permisos desde el entorno gráfico de Linux (Unity, gnome…) seleccionando el archivo, dándole al botón derecho del ratón, y luego a propiedades. O simplemente lo podemos ver si hacemos un ls -l y vemos que el archivo tiene los valores "x" donde debe tenerlos
· Desde Linux, abrimos una ventana de terminal, nos desplazamos a la carpeta que contiene el FastQC y lanzamos el programa FastQC invocando fastqc como $./fastqc
· Anotar que aunque estemos en la carpeta que contiene el archivo fastqc, hay que lanzar la orden dándole a “./”
· Y ya tenemos abierto el programa FastQC ya sea desde Windows o desde Linux

- Ya dentro de FastQC le doy al menú File y selecciono la carpeta que contiene los archivos descargados y en particular el archivo SR292770_1.fastq.gz del tirón, sin descomprimir. La navegación es simple, como en cualquier ventana de Windows.Y lo analiza. En poco tiempo te indica las estadísticas que ya hemos usado en la práctica anterior
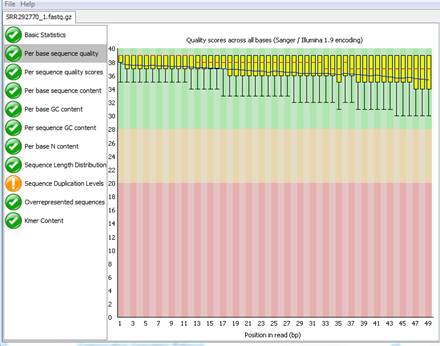
o Basic Statistics que se ha usado Illumina, versión 1,9, y que hay 5102041 secuencias con una longitud de 49 bases y un contenido de GC del 50%
o Per base sequence quality da una gráfica con las calidades pero a lo largo de las 49 bases que ha leído. Se puede observar una degradación de la calidad a lo largo del número de secuencia como es habitual debido al “Error de Fase” característico de este sistema de secuenciación. No obstante, el valior de Q es superior a 34 en todos los casos, implicando un porcentaje de acierto superior al 99,9%
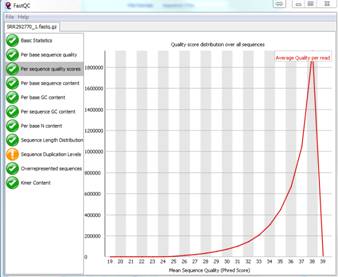
· En Per sequence quality scores una gráfica con las Q (Phred Scores) que me indica que el máximo es Q38 y se observa una distribución que indica que la calidad de la secuenciación es muy buena
· Los otros análisis son más irrelevantes en este caso porque no dan error, pero por ejemplo, se ha de comprobar en Per base sequence content, que hay el mismo número de A que T y de G que C, que la distribución de las longitudes secuenciadas es correcta.
· En Per base N content nos indica cuantas N aparecen en nuestra secuencias.
· En este caso FastQC destaca el apartado Sequence Duplication Levels que indica con una interrogación en color naranja (y no verde) que hay un problema, en este caso que hay un 26% de secuencias duplicadas. Pero en un principio no va a ser el propósito de esta práctica el tratar con ello.
· Desde el menú File/Save report se puede guardar los resultados del análisis. Guardo los datos del análisis desde la aplicación. Se puede ver que lo guarda archivos ZIP que contienen página WEB con imágenes que facilita su acceso posterior
· Abrimos el archivo SRR29770_2.fastqc.gz para realizar el mismo tipo de análisis
· Veo que el análisis es similar, que tiene la misma calidad. Guardo el report en la misma carpeta que el anterior.
· Hay muchas herramientas útiles para los sistemas NGS en http://omictools.com/quality-control/
CONTINUAMOS CON EL ENSAMBLADO
- Ahora nos metemos en el entorno de Linux.
- Aquí pueden surgir muchos problemas. Muchos
- Podemos no tener permisos para compilar programas si no somos root de dicho ordenador, esto es, si no es tu ordenador y tu lo has instalado convirtiendote en superusuario (su)
- Descargamos el programa Velvet desde su página WEB https://github.com/dzerbino/velvet/tree/master.
Hay un enlace en la parte derecha media que pone Clone or download. Si le das, verás que aparece la opción Download ZIP que te descarga
el programa
- Puedes hacer lo que hicimos en clase. Poner el ratón sobre el enlace que pone Download, darle al botón derecho del ratón, y entonces guardar el enlace del archivo
- Y a continuación, en una línea de comando escribes wget y le dar al botón derecho del ratón y Pegar
- Eso hará que aparezca un archivo zip en tu cuenta que puedes descomprimir con la orden unzip desde Linux. Te creará una carpeta que se llama velvet-master
-
Dentro de la carpeta verás que hay un archivo llamado Makefile. Ademas verás instrucciones y el manual del programa. En las instrucciones, verás que con la orden $make se debe compilar y generar los ejecutables.
Pero puede que la instrucción make no funciona por algún tipo de problema, como que no tengas instaladas las librerías zlib o cualquier otra librería necesaria (librería es en este caso alguna aplicación requerida adiconal al programa que estás usando). Si lees el mensaje de error verás que indica la causa del problema claramente. En estos casos de error verás que se han generado los archivos velveth y velvetg ejecutables. Para esto hay dos soluciones:
- Leer el manual del equipo que te dice como corregir este problema (ver Nota 1 abajo)
- O hacer una búsqueda en google con
“instalar zlib ubuntu” (o cualquier otra libreria que se indique en el mensaje con el problema). Cuando busco por "install zlib library Ubuntu" soy capaz de encontrar el modo de instalar estas librerías con la siguiente orden
$sudo apt-get install --reinstall zlibc zlib1g zlib1g-dev
Nota 1: Como alternativa, podemos ver que si me hubiera detenido a leer el manual de Velvet, se indica que podría haber compilado el programa con la orden
$./make ‘BUNDLEDZLIB=1’
Y eso hubiera instalado velvet con las librerías de compresión zlib que son necesarias. También hay otras opciones interesantes a la hora de compilar el programa, como la de indicar $make color para obtener unos ejecutables que manipulan las secuencias en la versión colorspace generado por el sistema de secuenciación SOLiD (en este caso se instalan otros dos archivos ejecutables con nombres diferentes). Otras opciones permiten manipular secuencias largas, usar más de un procesador simultáneamente, etc. Es cuestión de leerse el manual tranquilamente.
AHORA ES IMPORTANTE LEER BIEN EL MANUAL O LAS INSTRUCCIONES DEL PROGRAMA PARA CONFIGURAR EL Makfile de forma correcta
PRÁCTICA: Discutiremos en clase cuales son las órdenes que debemos cambiar o incluir en el Makefile ANTES de compilar y generar los ejecutables
Una vez que hayamos modificado el Makefile instalamos el programa Velvet con la orden
$./make
Nota 2: Si se usa el centro de software de Ubuntu, se puede encontrar velvet y se puede instalar de modo automático. Pero no lo recomendamos en este caso porque velvet se puede instalar con varias opciones diferentes, entre ellas, dándo a $./make color, que crearía una versión de velvet apta para su uso con el sistema color space del sistema de secuenciación SOLiD (que no veremos en las prácticas ya que el sistema SOLID ya no se usa) mas . Otras opciones son las de cambiar el máximo valor del k-mer que Velvet usará para generar las gráficas de Bruijn y que por defecto son 31 (se puede saber al ejecutar la orden velveth). Otras posibilidades están indicadas en el archivo Manual.pdf que se incluye dentro de la carpeta distribuida
(opcional) Dentro de la carpeta velvet que he descargado desde GitHub, hay un archivo que se llama Makefile. Los usuarios avanzados pueden editar y cambiar los valores de este archivo para cambiar las características de compilación y he cambiado el valor de MAXKMERLENGHT a 101, y he generado con Make los ejecutables. El valor de 101 es para manejar las secuencias de los nuevos secuenciadores MiSeq que son capaces de generar más de 100 bases
PRÁCTICA: No te fijes al 100% en esta imagen. Trata de incluir otras dos órdenes aquí en este Makefile (que puedes invocar con la orden $gedit Makefile para que Velvet sea capaz de usar más canales o categorias de secuencias y más de un núcleo de tu ordenador para acelerar el proceso del ensamblado

Y al final, compila el programa con make. Al compilar, se crean dos nuevos archivosv binarios (=programas) llamados velveth y velvetg, respectivamente. Ahora es el turno de hacer el ensamblado
NOTA IMPORTANTE: Hay una singularidad en el ordenador de la UCO. Aunque el sistema operativo Linux que tenemos instalado es de 64 bits (algo que se exige por parte de Velvet), el programa compilador (llamado gcc) que se ejecuta en la UCO por defecto es uno de 32 bits. Esto es así por que los ordenadores de la UCO se usan por miles de usuarios y hay que satisfacer todas las necesidades. Para arreglar este problema, y para que se use el programa gcc en su versión de 64 bits hay que ejecutar la orden source usegcc64 . (importante, no podemos olvidar poner el "." después de la orden usegcc64 para que los cambios tengan lugar en la misma shell (en la ventana de terminal) en la que estamos trabajando
Ahora tengo dos opciones para manejar cómodamente velvet, que si no, me voy a tener que meter los PATH en las órdenes y es una lata
OPCION 1. He metido temporalmente el PATH de los programas velvet con la siguiente orden. El PATH es la indicación de las carpetas donde Ubuntu busca los archivos y programas. Y no es como Windows, que una vez que instalas algo, se ejecuta sin más. En Ubuntu, si se instala un programa manualmente, hay que indicar donde está localizado (hay que indicar el PATH donde está la carpeta). Si escribimos:
$echo $PATH # esta orden indica el valor del PATH, o las carpetas o lugares en als que Linux busca los archivos y programas.
PATH="$PATH:$HOME/BioMol/velvet” # Esta orden añade el lugar donde esta velvet en el PATH de forma que Ubuntu lo encuentra (respeta los directorios.. pueden no ser los mismos que los que tu tienes en tu cuenta). Opcionalmente puedes ejecutar la orden export como en esta orden export PATH="~/BioMol/velvet:$PATH". Fíjate que en esta ocasión he usado la orden export, y que he sustituido el contenido de $HOME por la diéresis (AltGr + 4). Puedes buscar más información sobre la orden export en Google. Es importante conocer que este acceso se va a conceder en la shell en la que uses estos comandos, y por tanto no será una solución permanente, sino temporal. Cada vez que abras una ventana, deberás repetir esta orden.
OPCION 2. Ya que solo los programas velveth y velvetg son necesarios, he creado copias de ellos en una carpeta ya incluida en el PATH con esta orden (se necesita ser root para tener los permisos adecuados. Esto significa que lo puedes hacer en tu propio ordenador, pero no en el ordenador de la UCO)
$sudo cp velveth /usr/local/bin
$sudo cp velvetg /usr/local/bin
Esta segunda opción es más conveniente ya que es una solución permanente al copiar de forma permanente estos dos archivos en una carpeta que viene definida desde el principio por Ubuntu, en este caso la carpeta /usr/local/bin
Ahora, y desde la ventana Terminal, tengo que tener en cuenta qué carpeta contiene los datos de la secuenciación hecha por illumina. Te lo recuerdo con una orden que lista el contenido como $ls /usr/local/uco/bms/practica2. Esta carpeta con los datos se ha creado en un lugar del ordenador ts.uco.es de la UCO para que no ocupe el poco espacio que tienes en tu propia cuenta. Allí verás los archivos de las secuencias de Illumina SRR292770_1.fastq.gz y SRR292770_2.fastq.gz. Como puedes ver, son archivos comprimidos. Velvet los va a manejar sin problemas.
NOTA IMPORTANTE: Volvemos a tener singularidades si usamos el ordenador de la UCO y no en nuestro ordenador. EL uso de velveth genera archivos de gran tamaño que ocupan mucho espacio en nuestros discos. Y ya sabemos que tenemos el espacio de disco muy limitado en la UCO (no asi en nuestros propios ordenadores, donde todo lo que estoy comentando no es necesario que se realice). Para evitar problemas lo primero que vamos a hacer es crear una carpeta personal en un directorio especial de la UCO llamdo /tmp que tiene espacio de sobra porque su contenido es temporal y se borra todo al dia siguiente. Ejecutamos la orden mkdir /tmp/Nombre_de_usuario (por ejemplo, en mi caso mkdir /tmp/bb1rofra).
Ahora que conocemos donde estan los archivos de las secuencias (practica2), y hemos creado el archivo temporal para que guarde los datos de los nodos de las gráficas de Bruijn, ya podemos ejecutar la primera orden
Con la orden ./velveth (si no metemos la ruta que contiene este binario en el PATH) se ejecuta la orden y nos da la ayuda directamente (no hay necesidad de usar el cualificador -h o --help)
Si miramos la ayuda veremos que nos indica el uso de este programa de una manera similar a la que vimos con fastx-toolkit
Usage:
./velveth directory hash_length {[-file_format][-read_type][-separate|-interleaved] filename1 [filename2 ...]} {...} [options]
El significado de directory, hash-length y demás parámetros viene especificado a continuación en la misma ayuda, pero si sigues leyendo este documento verás que lo explico también aquí.
Y aquí tienes una orden que puedes ejecutar usando un valor de kmer para las gráficas de Bruijn de 35
$velveth /tmp/bb1rofra/Resul35 35 -fastq.gz -shortPaired -separate /usr/local/uco/bms/practica2/SRR292770_1.fastq.gz /usr/local/uco/bms/practica2/SRR292770_2.fastq.gz #Sustituir el bb1rofra por tu nombre de usuario
- /tmp/bb1rofra/Resul35 es el nombre de la carpeta donde va a guardar los resultados y los archivos intermedios. Como lo hacemos en el directorio tmp no tendremos problemas de espacio normalmente. Recordar que esta información se borrará al dia siguiente
- 35 es el valor de k-mer que hemos seleccionado para que genere las gráficas de Bruijn (el tamaño de los nodos que usará el programa). Si se mira la ayuda del programa, se puede ver que se puede definir tres valores m,M,s (como 15,41,2) en la que se definen nodos de las gráficas de Bruijn desde un tamaño de 15 bases a 41, saltando de dos en dos). Pero esto no lo vamos a hacer en esta práctica porque tardamos mucho más en hacerlo y se generan archivos mucho más grandes
- Con –fastq.gz le indicamos que use archivos fastq comprimidos como gz. Por eso necesitábamos las librerías zlib, que son librerías de descompresión
- Con –shortPaired le indicamos a velvet que tenemos secuencias pareadas y cortas. En este caso estamos usando un único canal, que es lo mismo que decir que estamos usando un solo tipo de secuencias
- Con -separate le estamos indicando al programa que los archivos de lecturas pareadas que vamos a usar están en dos archivos por separado, y no en uno con las secuencias entrelazadas (interleaved)
- Luego finalmente ya le damos el nombre de las dos secuencias que queremos procesar. Como veis se define la ruta completa que las contiene
En menos de 3 minutos, me ha creado con las 10204082 secuencias el RoadMap. En el Roadmap, velveth ha unido las secuencias pareadas que en un principio estaban separadas en los dos archivos. Para hacerlo ha buscado en las coordenadas de los archivos fastq. Y lo he hecho en la carpeta temporal que creamos /tmp/bb1rofra/Resul35.
PRÁCTICA: Lee el manual e indica cómo puedes ejecutar la orden anterior de tal forma que Velvet se ejecute con varios valores de K diferentes de tal forma que no tenga que volver a procesar todas las secuencias en cada uno de los pasos
Ahora ejecuto la parte de velvet que genera el ensamblado
$velvetg /tmp/bb1rofra/Resul35 -clean yes -exp_cov 21 -cov_cutoff auto -min_contig_lgth 200 # Recuerda que donde pone bb1rofra, has de poner el nombre de tu cuenta
Y el programa se pone a ejecutarse buscando los consensos. Velvetg es el programa que genera y usa las gráficas Bruijn, que señala y busca los nodos, que los une para ensamblar y que termine creando contigs (continuidades ) y scaffoldings, que concatena…
- /tmp/bb1rofra/Resul35 hará que velvetg busque el Roadmap en la carpeta Resul35 que fue creada por velveth
- -clean yes borra todos los archivos intermedios que crea durante el análisis. Eso nos permite ahorrar espacio en disco. Esto es necesario hacerlo solo en la Universidad de la UCO por los problemas de espacio que tenemos
- -exp_cov 21 es un modo de indicarle a Velvet cuál es el tamaño esperado de nuestro genoma indicando la cobertura que tenemos. Pero en esta ocasión la cobertura que se indica, no es la cobertura a nivel de bases, sino la cobertura que Velvet usa que es la de los subgragmentos con el kmer que hemos seleccionado. Este valor se calcula con la formula Ck = C (L-k+1)/L, donde C es el valor de cobertura tradicional a nivel de bases (ya sabes, si nuestro genoma es de 1Mb, y hemos secuenciado en total 2Mb, este valor C es de 2). L es la longitud de las secuencias, en nuestro caso 49 bases. k es la longitud de los k-mer que hemos seleccionado, en este caso 35. Esta información la he sacado del manual del programa
- -cov_cutoff auto determina que velvetg va a eliminar del análisis muchos nodos que proporcionan secuencias con baja cobertura de secuenciación de una forma automática. En el manual de Velvet se indica el modo en el que nosotros mediante un paquete del programa R podemos averiguar ese valor. Esto lo tengo indicado un poco más adelante en este mismo documento.
- -min_contig_lgth obliga a velvetg a no considerar aquellos contigs que tengan menos de 200 bases
Para calcular el valor de cov_cutoff manualmene, podemos usar R para sacar cierta información usando el archivo stats.txt que se genera con velvetg
BiocManager::install("plotrix") #Esto instala el paquete plotrix
library(plotrix) # Carga el paquete
x11() # Abre una ventana gráfica con las imágenes que se van a generar
data <- read.table("stats.txt", header = TRUE) # Lee la tabla e incluye los nombres de las columnas
weighted.hist(data$short1_cov, data$lght, breaks= 0.50) representa los datos
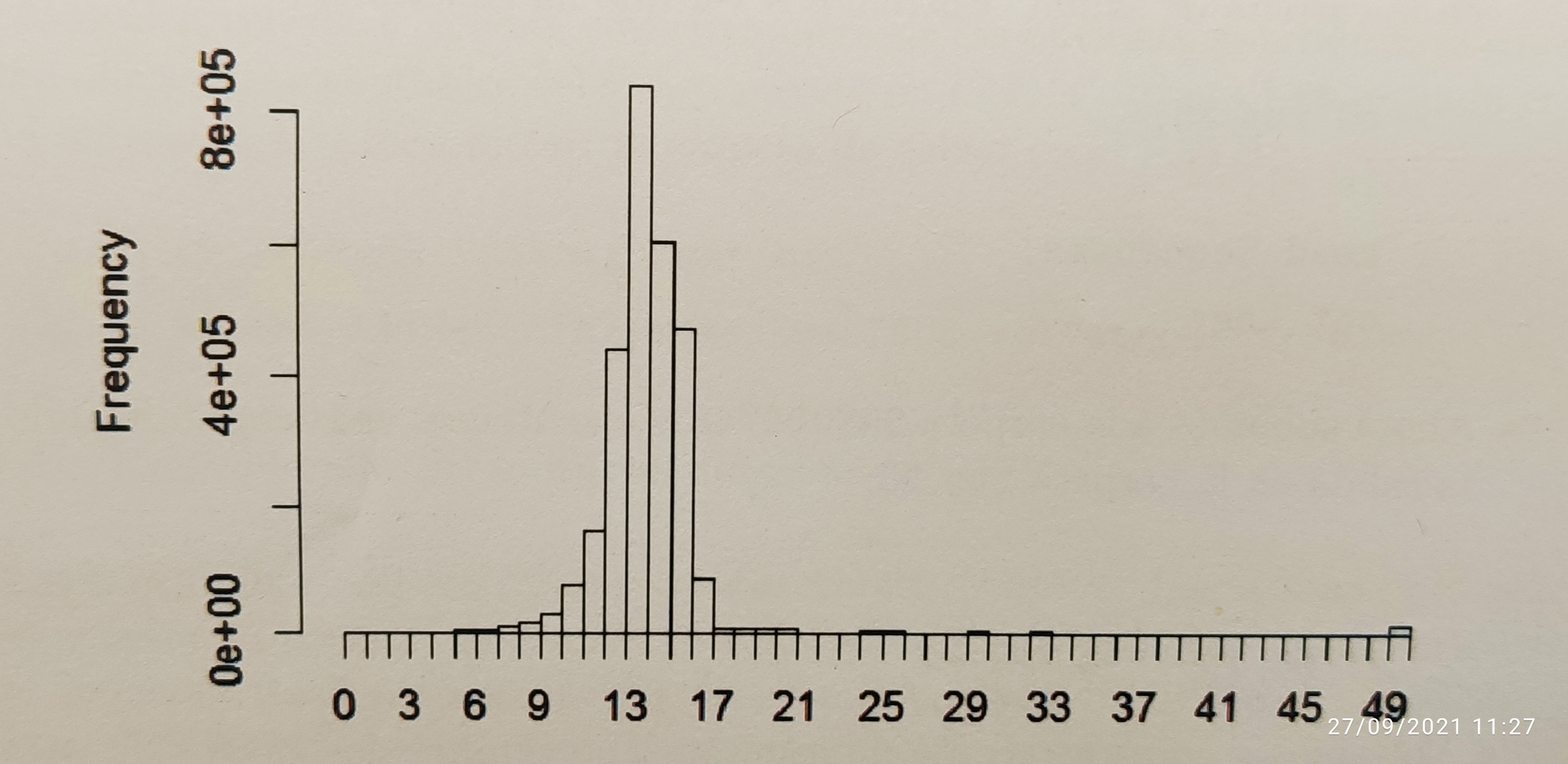
De la imagen proporcionada por el análisis con R se puede obtener la información de la cobertura. Así, en este caso, se ve que se espera una cobertura alrededor de 14 y que todo lo que tenga una cobertura inferior a 6, podria ser ruido. Las coberturas que se observan alrededor de 30-35 o mayor de 50 pueden corresponder a secuencias repetidas
Según este gráfico, velvetg se podría ejecutar más optimizado con la opción cov_cutoff 6 exp_cov 14
ALTERNATIVAS PARA OBTENER INFORMACIÓN DEL ENSAMBLADO
Si uno lee el manual, verá que hay un formato especial de los archivos resultados que se pueden proporcionar en el llamado formato amos. Amos es un programa de visualización ciertamente antiguo que permitía ver el conjunto de lecturas que velvetg ha ensamblado. Nosotros usaremos un programa más moderno y más adecuado porque está realizado en java y se puede ejecutar en las plataformas Windows, Linux y MacOS. Se llama tablet, y se puede obtener desde este enlace https://ics.hutton.ac.uk/tablet/download-tablet/
ay más opciones que se pueden usar con velvet que aparecen con solo lanzar $velvetg | more. Una de ellas es la opción amos_file yes que genera los archivos convencionales y, ademas, el archivo en formato amos que el programa tablet puede leer y presentar sin problemas. Para ello, lo único que hay que hacer es ejecutar velvetg con las órdenes de comandos deseadas, y añadir el cualificador amos_file yes. De esta forma se generará un archivo con la terminación afg (Amos Format Graphic) que se abre directamente en Tablet.
Tablet nos proporciona imágenes como ésta
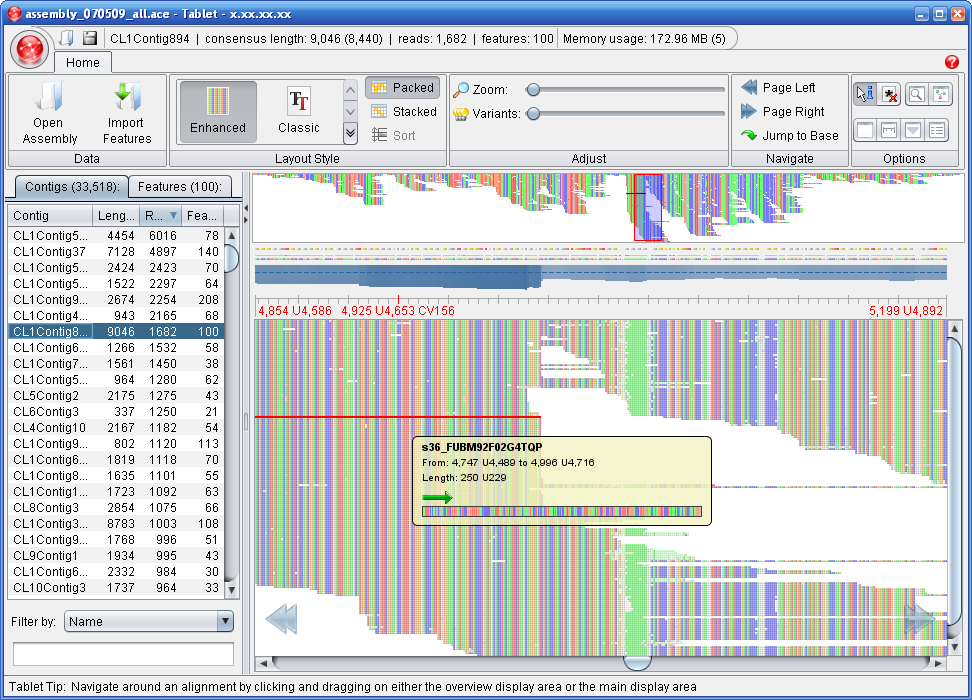
Aquí se ven a un nivel de zoom elevado cómo se han alineado las distintas lecturas para generar el consenso en el ensamblado
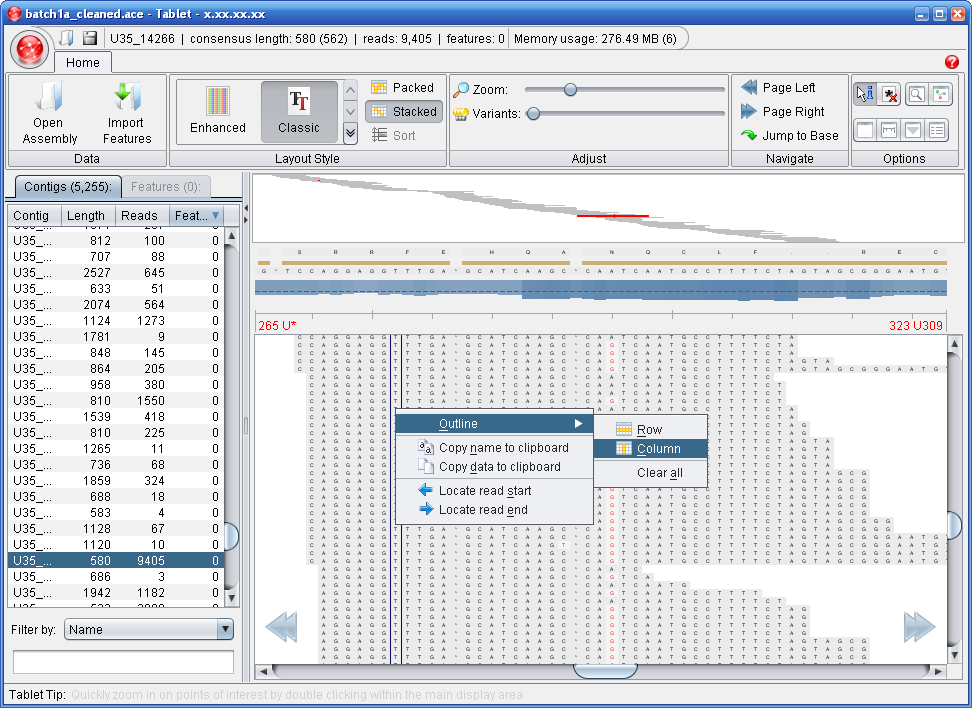
Y aquí se ve la misma información pero llegando al nivel de las bases
CUIDADO con la orden de comandos. Respetar los parámetros, no poner símbolos igual, no errar a la hora de escribir los cualificadores porque el programa no los entendería, dando errores de forma continuada respetad las mayúsculas y minúsculas. En otras palabras. En la medida de lo posible tratar de usar el tabulador como hemos indicado en clase.
En unos 195 segundos ha acabado.. Tenemos una propuesta de la secuencias ensamblada de novo de E.coli que está dividida en scaffolds. Los archivos que necesitamos son contigs.fa (los scaffolds) y si acaso stats.txt. Los demás archivos los podemos borrar para ahorrar espacio.
· Ahora conviene considerar que el ensamblado va a depender del valor de K que le demos a velvet en el momento generar los nodos para su consideración por velvetg a la hora de crear las gráficas de Bruijn
· Una opción es lanzar velveth y velvetg manualmente con diversos valores de k (lee el manual para ver como se hace con una sola orden desde velveth)
· Pero otra opción es la de usar scripts que lanzan los programas velveth y velvetg de forma automática y que están incluido en una de las carpetas que hemos obtenido al descargar Velvet. Vamos a ver primero el programa Velvetoptimizer.pl que está dentro de la carpeta contrib del propio velvet. Este programa está escrito en Perl, y Perl está instalado por defecto en Ubuntu
EJECUTANDO ALGUNOS DE LOS PROGRAMAS QUE SE INSTALAN EN LA CARPETA CONTRIB (avanzado, voluntario)
Ahora me propongo instalar VelvetOptimizer y asegurarme de tener instalado todas los programas y dependencias que requiere como Bioperl. BioPerl es un conjunto impresionante de rutinas de interés para los bioquímicos y biólogos moleculares que puede funcionar bajo Linux, Windows, Mac OSX, Ubuntu, FreeBSD y Fedora
Considerar que hay otras versiones equivalentes a BipPerl, como BioPhyton o BioJava..
$perl –version
me indica que tengo la versión de Perl 5.14. El tutorial me dice que por encima de 5.8 vale. Así que lo dejo
Ahora tengo que instalar BioPerl
· Puedo optar por leer la documentación incluida en http://www.bioperl.org/wiki/Main_Page
· También puedo buscar bioperl usando el icono de Centro de Software de Ubuntu
· También para facilitar la instalación de BioPerl y otros programas puede instalar el programa Synaptic en Ubuntu, una pequeña maravilla..
$sudo apt-get install synaptic
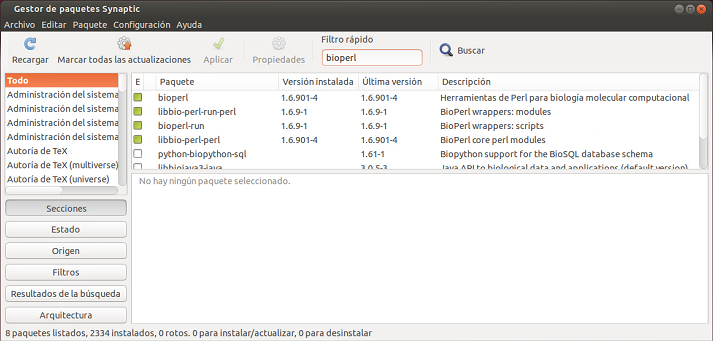
Abro el programa Synaptic usando el icono lateral “Busca en su equipo y en línea” , y en el campo de búsqueda escribo y busco BioPerl. Le pido que me lo instale, y lo hace no sin tardar un poquito. Synaptic facilita la instalación de programas y librerías y tiene en cuenta todas las depedencias (programas y archivos adicionales) que se requieren para el correcto funcionamiento de los programas. Además Synaptic te instala las librerías en los lugares adecuados para que se incluyan en el PATH y así sean capaces de ejecutarse de forma adecuada
Ahora descargo VelveOptimiser 2.2.5 y lo ejecuto con la siguiente orden (no hay que instalar nada, ya que Velvetoptimizer es un archivo ejecutable escrito en Perl
$./Velvetoptimiser.pl -s 35 -e 41 -f ‘-fastq.gz -shortPaired /ruta/SRR292770_1.fastq.gz /ruta/SRR292770_2.fastq.gz’-o ‘-min_contig_lgth 200’ -p SRR292770
Esta orden está indicando en que ejecute 4 órdenes consecutivas de velveth con valores de kmer 35, 37, 39 y 41 (recordar que cuando se usan gráficos de Bruijn hay que poner valores impares de k). Después, el mismo script lanza velvetg cuando se han generado los Roadmaps. Luego toma el resultado con el mejor N50 como la semilla para la optimización final. Todo lo mete en una carpeta nueva llamada SRR292770-data-XX, donde XX es el valor de k-mer elegido para que no se confunda con la anterior
· En la carpeta contrib de velvet se encuentran otros programas útiles, como AssemblyAssembler que es un programa escrito en phyton que hace algo similar a VelvetOptimizer: lanza automáticamente velvet con varios valores de K, pero luego es capaz de trabajar con los contigs que se crean con cada valor de k para intentar hacer un ensamblado final. Para que funcione AssemblyAssembler es necesario tener instalado phyton en su versión 2.X (no es válida la versión 3.X). Se recomienda la instalación de la versión de Phyton de ActiveState que se puede descargar desde http://www.activestate.com/activephyton
· Entramos en activephyton y le damos al botón que pone Download Now, y se descarga la versión Phyton 2.7.5. Hay versiones de pago que tienen soporte, pero nosotros vamos a descargar la versión gratuita sin dicho soporte. El archivo tiene unos 30,9Mb de tamaño en formato tar.gz. EL sistema Ubuntu trata estos archivos automáticamente sin más que darle un doble clic sobre el una vez descargado para que lo descomprima (gz) y para que separe los archivos y carpetas (un-tar)
· Entramos en el directorio ya descomprimido y “untar-trado” y escribimos $sudo ./install.sh desde una ventana de terminal (recordar que hay que tener permiso de root para poder hacerlo). De esa forma instalamos phyton en los directorios declarados por el programa por defecto
· Una vez instalado phyton podemos ir al directorio velvet/contrib/AssemblyAssembler y podemos ejecutar como comando la orden $./AssemblyAssembler1.3.py. En dicha carpeta viene un documento README.txt que indica cuales son los parámetros exigidos para que funcione. También indica que se le debe de dar permisos al ejecutable con la orden $chmod 777 AssemblyAssembler1.3py
· Por comodidad, yo he renombrado el archivo de esta forma $mv AssemblyAssembler1.3py AA1.3.py
· Un ejemplo de cómo pueden ser estas órdenes vienen en el propio README.txt, un archivo dentro de la carpeta Assembly Assembler
· Podéis optar por dar la orden $sudo cp AssemblyAssembler /usr/local/bin para tenerlo en el PATH y poder ejecutarlo desde cualquier directorio como ya hemos hecho con otros comandos.
· EL uso de AssemblyAssembler puede tener interés ya que como hemos indicado, no solamente lanza velveth y velvetg con diferentes valores de k-mer, sino que tratará de usar los archivos contig.fa creados en cada caso (son los contigs y scaffolds) para generar un nuevo consenso. Realmente habría que valorar como lo hace. Mi sugerencia es que lo realicéis como una práctica adicional. Leeros el archivo README.txt de la carpeta o directorio AssemblyAssembler1.3 para ver cómo funciona. Ese documento contiene varias órdenes para ver como lanzarlo
· Ya sea de una forma o de otra, con ayuda de programas como VelvetOptimizer, o AssemblyAssembler, hemos terminado de ensamblar las secuencias con varios valores de k. Por cada valor de K, se ha creado una carpeta diferente, y dentro de cada carpeta hay un archivo contigs.fa que contiene los contigs y scaffolds que velvetg ha generado
· Ahora es el turno de evaluar cuál es el mejor valor de k. Una opción es la de usar un genoma de referencia de E.coli ya publicado, y luego, con ayuda de un programa capaz de alinear nuestros contigs y scaffolds, valorar cuál es el que mejor resultado o similaridad tiene.
· Para este fin vamos a usar el programa Mauve
ORDENANDO LOS SCAFFOLDS CON AYUDA DE MAUVE
· Descargamos el paquete Mauve desde http://darlinglab.org/mauve/mauve.html
· Se trata de un programa con ventana gráfica GUI que se ejecuta bajo JAVA. La utilidad de este programa es múltiple. Crea consensos alineando genomas completos, mueve fragmentos del genoma, une scaffolds..
· Buscamos la parte que indica Download Mauve
· Seleccionamos el sistema operativo Linux del campo desplegable y le damos a Download, pudiendo o no incluir el nombre o E-mail
· Se descarga un archivo comprimido pero se descomprime de forma habitual con el propio Ubuntu (basta hacer un doble clic sobre él)
· Yo lo he creado en la carpeta biomol/mauve. Dentro de ella hay un archivo llamado Mauve.jar que se puede ejecutar con la orden (ojo con las mayúsculas..)
$java -jar Mauve.jar

· Antes vamos a bajar un genoma de E.coli que usaremos como genoma de referencia. Para ello entramos en https://www.ncbi.nlm.nih.gov/nuccore/NC_011748.1
· Desde esa página descargamos el archivo dándole a la opcion Send To / File / formato Fasta y lo guardamos como NC_011748.fna y lo guardamos en la misma carpeta donde tenemos el tutorial. Este es el genoma de esta estirpe de E.coli en formato fasta. Yo lo he renombrado como Ecoli55989.fa con la siguiente orden $mv NC_011748.fna Ecoli55989.fa
· Si habéis tenido algún problema con velvet, y no habéis podido hacer el ensamblado vosotros, podéis descargar los archivos ya ensamblados de esta estirpe de E.coli desde este enlace http://www.ncbi.nlm.nih.gov/Traces/wgs/?val=AFVS01. Debéis pulsar sobre la pestaña Download y descargar el archivo Fasta denominado AFVS01.fsa.1.gz de 1,7Mb. Lo descomprimis directamente desde una ventana gráfica de Ubuntu con un doble clic
· Nos centramos en el programa Mauve. Entramos en el menú Tools, y seleccionamos Move Contigs. Una ventana de ayuda pequeña te indica que vas a ordenar contigs
· Se abren tres ventanas. Una ventana indica “Choose location to keep output files and folders” que es donde se van a guardar los resultados del ordenamiento hecho por Mauve. Yo he creado una carpeta llamada ana_mauve dentro de la carpeta BMS

· Creamos la carpeta donde van a ir instalados los resultados de mauve y buscamos ahora otra ventana que nos va a permitir introducir las secuencias que queremos comparar (nota: hay una tercera ventana, Mauve Console que esconde a la que te deja meter las secuencias. Muevela para ver la que necesitas que se queda escondida abajo).
· Ahora mismo lo que vamos a hacer es meter la secuencia de referencia Ecoli55989.fa (el genoma de referencia que he guardado en la carpeta ana_mauve), y el archivo contigs.fa que ha generado velvetg dentro de la carpeta /tmp/bb1rofra//Resul35 (en realidad se debería realizar varios análisis de velvet con varios valores de k y aportar aquella secuencia procesada por velvetg que tenga el valor de N50 más grande)
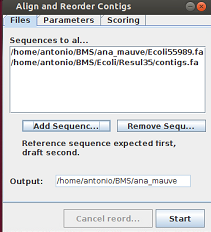
· Una ventana nos anunció inicialmente que nada mas que le demos a Start va a empezar a hacer un trabajo reiterativo que puede tardar tiempo, así que cuidado.. En la ventana Mauve Console podemos ver el desarrollo del proceso de Alineamiento y reordenamiento de los contigs conforme este se va desarrollando
· ESCOGER PRIMERO EL GENOMA DE REFERENCIA desde la ventana que se abre, dándole al botón Add Sequence. Así sabe el programa que no debe reorganizar ese genoma, sino el que indiquemos en segundo lugar
· Darle al botón Start. El programa puede tardar más de media hora en hacer el trabajo. Se abre una consola donde se puede ver cómo evoluciona el trabajo. El reordenamiento puede tomar desde 4 a 13 interacciones dependiendo del sistema operativo (en Mac hasta 16 interacciones.. e incluso más en un Windows de 32 bits)
· EL programa va mostrando ventanas con los diferentes alineamientos. Con cada propuesta de alineamiento, presenta una nueva ventana
· El resultado de Mauve se va a mostrar en la carpeta de resultados donde se han creado varias carpetas, cada una de ellas con un alineamiento distinto. El resultado final es el que está en la carpeta alignmentX donde X es el mayor número. En mi caso :
o ha hecho 13 alineamientos en el VirtualBox del ordenador de mi casa bajo Windows 7 64bits… La conclusión es evidente. Los dos genomas que hemos secuenciado no se parecen entre si
o En el Servidor Ubuntu de la Universidad ha hecho de 8 a 10 alineamientos, con un resultado bastante variable dependiendo del numero de k usado
· Ahora os muestro las diferencias en los alineamientos hechos cuando se ha usado un k-mer de 25, 31 o 35 a la hora de ensamblar con velvet
· Estos alineamientos con diferentes valores de k nos permiten valorar cual es el ensamblado más adecuado. Es conveniente incluso hacer cambios en los parámetros del ensamblado (como valores de cut_off, etc, hasta conseguir el mejor resultado
Abajo veis el resultado cuando se ha usado un valor de K= 25 (ha requerido 4 alineamientos). Se generan 773 LCB o uniones

Los bloques de colores representan segmentos de secuencias de DNA que son similares entre el genoma de referencia (arriba) y el ensamblado que hemos realizado (abajo). Las líneas cruzadas de la parte media representan donde están localizadas zonas más o menos comunes reconocidas por el programa. Podemos jugar con el valor de zoom para ver más detalles. Si os fijais, se indica de cierto modo qué cadena de ADN (si la + o la -) se corresponden entre si
Abajo veis el resultado cuando se ha usado un valor de K= 31 en velveth (ha requerido 10 alineamientos ) (Nota: el ensamblado se ha hecho con los mismos parámetros, solo se ha cambiado el valor de K). Aquí se observa que el genoma es más parecido y que solo se han formado 183 LCB
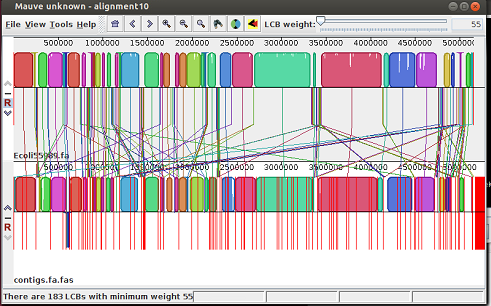
Observar la mucha mayor calidad en el ensamblado que se ha obtenido con el valor de k=31 frente al obtenido con el K= 25. Los bloques en colores son bloques creados por el programa que tienen homología entre las dos secuencias. Las secuencias en rojo corresponden a secuencias presentes en contigs.fa que no tienen correspondencia con el genoma de referencia
Vamos a valorar qué ocurre cuando aumentamos el valor de k un poco más
Abajo veis el resultado cuando se ha usado un valor de K= 37 (ha requerido 8 alineamientos)

En este último caso vemos que se ha vuelto a deteriorar la calidad del ensamblado
· Está clara la influencia del valor de K en el ensamblado y en el reconocimiento por parte de mauve de secuencias comunes entre nuestro ensamblado y en el del genoma de referencia. Obviamente no estamos controlando la calidad del genoma de referencia que estamos usando. Por ello, es muy recomendable realizar un estudio comparativo con más de un genoma de referencia y/o ensamblado, incluso con aquellos ensamblados que se han conseguido usando otros programas, para buscar aquella combinación empírica que nos dé mejores resultados. Eso lo podemos hacer también con mauve
· En este caso la calidad del ensamblado usando K= 31 es la mejor de todas por ahora. Los bloques de homología con la secuencia de referencia son los más grandes. El número de líneas que conectan secuencias similares pero que no están en el mismo orden o en la misma cadena (cadena + o -) es mucho mejor. Mauve ha encontrado el ensamblado que más se aproxima al del genoma de referencia. Quizás se debería reanalizar los datos con un valor de K=29 y un K=33 buscando si hay mejora o no en el alineamiento. EL propio programa mauve nos da el valor de LCB (Lines connecting Homologous sequences)
Abajo veis el resultado cuando se ha usado un valor de K= 33 (ha requerido 4 alineamientos). EL valor de LCB (195) es muy similar al obtenido cuando K=31 (183).

Así que damos por óptimo el valor de 31 aunque aún no hemos realizado este análisis con el valor de k = 29. Es una buena oportunidad para que lo hagáis como práctica
NOTA: Y que Mauve nos ha organizado, ordenado y mejorado el ensamblado al manipular los contigs, ahora podemos usar esta nueva información. Más detalles en esta página http://darlinglab.org/mauve/user-guide/reordering.html
REORGANIZANDO Y VIENDO LOS CONTIGS ORDENADOS DE OTRO MODO..
· Vamos a hacer dos aproximaciones
o Con Mauve, vamos a generar un alineamiento múltiple con el ensamblado que hemos hecho, con la secuencia del genoma de referencia Ecoli55989 que nos hemos descargado desde el NCBI, y con una tercera secuencia ensamblada con más secuencias que las que hemos usado para ensamblar nuestra secuencia y que ha sido ensamblada con otro ensamblador. Se trata de la estirpe TY-2482 (Numero de accesión NCBI AFVR01) que se puede descargar desde http://www.ncbi.nlm.nih.gov/Traces/wgs/?val=AFVR01. Descargamos desde la pestaña Download el archivo AFVR01.fsa.1.gz de 1.6Mb que descomprimimos en donde están los demás archivos
o Ahora le damos al menú de Mauve File, Align with progressive Mauve
o Se abre una ventana para añadir secuencias
o Añadimos un total de 3 secuencias que introducimos por este orden: La secuencia que procedente de nuestro ensamblado con velvet, fue organizada por Mauve con un K=31 y que se encuentra localizada con el nombre contigs.fa.fas dentro de la carpeta ana_mauveK31/alignment10, luego la secuencia de E.coli de referencia NC_011748 y luego la nueva secuencia AVFR01.fsa. En el caso del servidor Ubuntu este es el orden que le he puesto
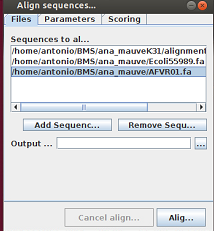
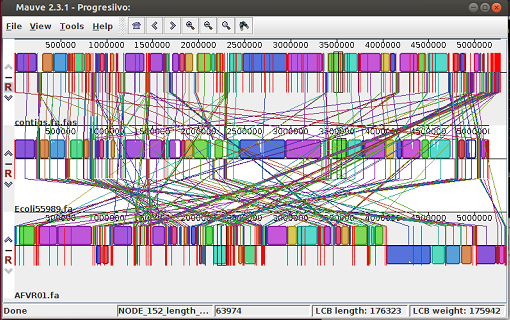
· Le damos al botón Align y esperamos el resultado que nos da
Si le doy a View, Style y le quito LCB connecting lines, se quitan las líneas y se puede ver algo más y mejor
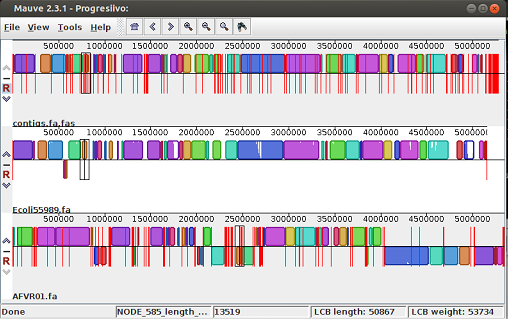
· Los resultados indican que nuestra bacteria está más próxima al genoma de referencia que hemos usado que lo es la bacteria con la que se ha conseguido el ensamblado AFVR01.fa. SI nos movemos con el ratón por los bloques en mauve, podremos ver como los bloques entre las 3 bacterias se conservan en mayor o menor grado, pero existe una regorganización génica importante en AVF01.fa cuando se usan los dos genomas anteriores como referencia
· Esta información nos sirve para corroborar cuál es el mejor valor de K para hacer el ensamblado
COMPARANDO GENOMAS CON ACT
· ACT es un programa creado por el centro SANGER que permite hacer una comparación gráfica de genomas completos. Exige la introducción de secuencias completas en formato fasta. No admite introducir secuencias divididas en contigs y scaffolds. La diferencia con mauve es que ACT no es capaz de comparar las secuencias y tampoco de reorganizar los contigs. Se limita a comparar dos genomas
· Entro en la página WEB http://www.sanger.ac.uk/resources/software/act/ y descargo la versión adecuada de ACT, que funciona bajo un entorno java gráfico. Se descarga un archivo tar.gz que se descomprime en una carpeta llamada Artemis. En esa carpeta Artemis, veremos dos archivos ejecutables ACT (que lanza ACT) y ART (que lanza Artemis, otro programa de utilidad)
· Entro en $cd biomol/Artemis desde una ventana de Terminal
· Me leo el manual y veo que lo que he descomprimido para UNIX (equivalente a Linux) ya está preparado para funcionar. Con un $ls -l veo que hay al menos dos archivos ejecutables llamados act (ACT) y art (Artemis).
· Lanzo Artemis con la orden
$./art

· Ahora navego con el File / Open y cargo el archivo fasta que fue organizado previamente por mauve, y que en el servidor Ubuntu está en la carpeta BMS/ana_mauve y que yo he llamado Alig10_mivelvetk31.fa

· También es posible obtener el fasta completo si entramos en las carpetas generadas por mauve. Dentro de ellas está el archivo contigs.fa que contiene la propuesta creada por Mauve
· ACT, que compara genomas, no puede funcionar con archivos fasta que estén separados. Es por ello que hemos abierto Artemis. Ahora debemos crear un único archivo fasta con todas las secuencias. Lo hacemos con la orden File / Write / All bases / FASTA Format. Le he dado el nombre Total_fasta_Alig10_mivelvetk31.fa
· Ahora hay que hacer un archivo de comparación para que funcione en ACT. Entro en synaptic y busco blast. Lo selecciono y lo instalo
· Abro una ventana Terminal y me voy a $cd BMS/ana_mauve que es donde tengo el archivo Total… que acabo de crear con Artemis
· Lanzo el blast pero desde la ventana de comando. Primero creo una base de datos local con el archivo del genoma de referencia
$makeblastdb -in Ecoli55989.fa -dbtype nucl
· Ahora lanzo el blast localmente con mi secuencia Total…
$blastn -query Total_fasta_Alig10_mivelvetk31.fa -db Ecoli55989.fa -evalue 1 -task megablast -outfmt 6 > Compara_miseq_referencia.blast
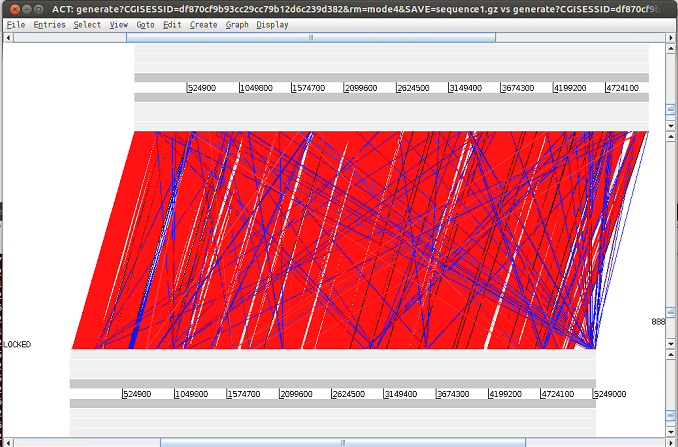
· Alternativamente, se puede accede a un servidor WEB para hacer dicha comparación http://www.webact.org. Se abre la página, se le da Generate y se puede cargar los dos archivos. Primero cargo el genoma de referencia, y luego el Total.. usando los botones de Examinar.. EL programa tarda un poco en cargar los archivos y un poco más en procesarlo.. Te ofrece un link en el que se guardan los datos durante 7 dias. Lo bueno es que tras darte los resultados, puedes descargar los datos en formato ZIP y lanzar ACT directamente desde esa página. El archivo zip contiene los fasta de los dos archivos usados y el archivo de comparación que se necesita
· Alternativamente se puede usar la página http://www.hpa-bioinfotools.org.uk/pise/double_act.html y accediendo a browse se hace básicamente lo mismo
REALIZANDO MÉTRICA DE ENSAMBLADO CON mauve
Mauve admite la instalación de una serie de script que analiza los ensamblados que se han realizado. Se ha comprobado que funciona en Macintosh, y vamos a ver si funciona en Linux
Hay que entrar en esta página
http://code.google.com/p/ngopt/wiki/How_To_Score_Genome_Assemblies_with_Mauve
En realidad, esta página WEB ofrece un método en el que se usa una estrategia basada en el que se tiene un genoma de referencia de alta calidad que se parezca lo bastante a nuestros genomas, y como se pueden comparar y usar genomas ensamblados con varios programas y algoritmos para conseguirlo. EL programa sirve para genomas haploides y poliploides homozigóticos, pero no por el momento para organismos diploides y poliploides divergentes
La página que describe este proceso da dos alternativas descargables desde
http://ngopt.googlecode.com/svn/trunk/tools/mauve_assembly_metrics/
· Un archivo para su uso con R de nombre mauveAssemblyMetrics.R
· Un archivo perl de nombre mauveAssemblyMetrics.pl
Yo descargo el archivo perl (.pl) y lo guardo en la carpeta biomol/mauve
Si quisiera descargar y usar R podría instalarlo con la orden
$sudo apt-get install r-recommended
Visitad la página y mirad la parte que pone A complete example of the Linux command line
· En el ejemplo, el autor descarga los archivos mauveAssemblyMetrics.R y pl con la orden wget, los “un-tar”, y con la orden $sudo chmod 755 mauveAssemblyMetrics.* hace ejecutable estos dos archivos. Al mismo tiempo descarga un archivo de referencia en formato gbk de Haloferax, y al menos 3 ensamblados distintos que ha conseguido
·
Leer toda la pagina donde se indica como se puede usar mauve bajo
comandos para dar toda la métrica
ANOTACION DE SECUENCIAS
Los genomas bacterianos y de arqueobacterias se pueden anotar automáticamente con un servidor llamado RAST (Rapid Annotation using Subsystem Technology) que se puede encontrar en http://rast.nmpdr.org. El servicio es gratuito pero exige que nos registremos..
· El input que necesita el programa es un archivo ordenado de contigs (en formato multi-fasta)
· Te registras en tu cuenta, entras, le das al botón “Your jobs” y se selecciona “Upload New Job”
· Se siguen las instrucciones para cargar el archivo ordenador, en nuestro caso Alig10…
· Luego hay que introducir la información taxonómica en el apartado “Review genome data”. Hay que introducir un código para que obtenga esa información. Si no conoces la taxonomía de tu organismo, la puedes comprobar en la pagina http://www.ncbi.nlm.nih.gov/taxonomy
· Se rellenan todos los demás datos aunque no es estrictamente necesario, y al final se le da a “Finish the upload”.
· La secuencia se carga en el servidor y se procesa en una cola de trabajo. Cuando el trabajo esté terminado (puede tardar media dia o incluso mas) se manda un correo para avisarlo
· En cualquier momento puedes entrar en la página para ver tus trabajos y cuál es el progreso del mismo
Alternativas a RAST
· Hay una serie de herramientas para poder anotar en una máquina local con Prokka que se puede obtener desde http://www.vicbioinformatics.com/software.prokka.shtml. Solo hay que seguir las instrucciones e instalar todo el resto de programas que se indica allí
· También se puede hacer una anotación comparativa con BG7 accediendo a http://bg7.ohnosequences.com
COMPARACION DE MULTIPLES GENOMAS CON BRIG
El programa BRIG permite comparar varios genomas diferentes en un entorno gráfico bajo JAVA
· Se puede descargar el programa desde http://brig.sourceforge.net que incluye un tutorial que puede ser útil
· BRIG requiere tener instalado BLAST, que o bien se instala a través de Synaptic o desde ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST
En esta página hay un tutorial de BRIG
http://brig.sourceforge.net/brig-tutorial-1-whole-genome‐comparisons/
· Lo que se ha de hacer es cargar un montón de secuencias de E.coli y la nuestra para comparar. Se puede incluso cargar archivos con anotaciones que incluya por ejemplo genes de patogenecidad para marcar donde están, etc..

Y es que este es lo que pasa.. Que no hay como es obvio un solo tipo de ensamblado para una misma bacteria aunque se la suponga tan simple como E.coli. Aquí tenéis un dendrograma de un montón de variedades que justifica las variaciones que hasta ahora hemos observado.

Las conclusiones
1. Que debemos ensamblar con un ensamblador de garantía bajo varias condiciones diferentes, o incluso valorar la posibilidad de ensamblar los mismos datos con diferentes ensambladores. Cabe la posibilidad de usar programas de terceras partes como AssemblyAssembler si se usa Velvet
2. Luego hay que buscar no uno, sino varios genomas de referencia con los que inicialmente haremos una comparación para ordenar nuestros contigs
3. Luego debemos valorar cual es el mejor ensamblado por ser el que mejor o más semejanza presente con un genoma de referencia. El simple hecho de que nuestra secuencia se parezca al del genoma de referencia valida los datos del propio genoma de referencia, porque éste ha sido ensamblado con unos datos y tecnología diferentes
4. Desde ya asumimos que nuestro ensamblado no va a ser perfecto. La comparación con mauve y sobre todo si conseguimos su métrica, nos va a indicar que porcentaje de ese genoma ensamblado es útil